The re-sampling-based null distribution is almost identical to the theoretical null distribution, which is displayed in red.
Finally, we implement the plug-in re-sampling FDR approach outlined
in Algorithm 13.4. Depending on the speed of your computer, calculating
the FDR for all 2,308 genes in the Khan dataset may take a while. Hence,
we will illustrate the approach on a random subset of 100 genes. For each
gene, we first compute the observed test statistic, and then produce 10,000
re-sampled test statistics. This may take a few minutes to run. If you are
in a rush, then you could set B equal to a smaller value (e.g. B = 500).
m <- 100
set.seed(1)
index <- sample(ncol(x1), m)
Ts <- rep(NA, m)
Ts.star <- matrix(NA, ncol = m, nrow = B)
for (j in 1:m) {
k <- index[j]
Ts[j] <- t.test(x1[, k], x2[, k], var.equal = TRUE)$statistic
for (b in 1:B) {
dat <- sample(c(x1[, k], x2[, k]))
Ts.star[b, j] <- t.test(dat[1:n1], dat[(n1 + 1):(n1 + n2)], var.equal = TRUE)$statistic
}
}
Next, we compute the number of rejected null hypotheses \(R\), the estimated number of false positives \(\widehat V\) , and the estimated FDR, for a range of threshold values \(c\) in Algorithm 13.4. The threshold values are chosen using the absolute values of the test statistics from the 100 genes.
cs <- sort(abs(Ts))
FDRs <- Rs <- Vs <- rep(NA, m)
for (j in 1:m) {
R <- sum(abs(Ts) >= cs[j])
V <- sum(abs(Ts.star) >= cs[j]) / B
Rs[j] <- R
Vs[j] <- V
FDRs[j] <- V / R
}
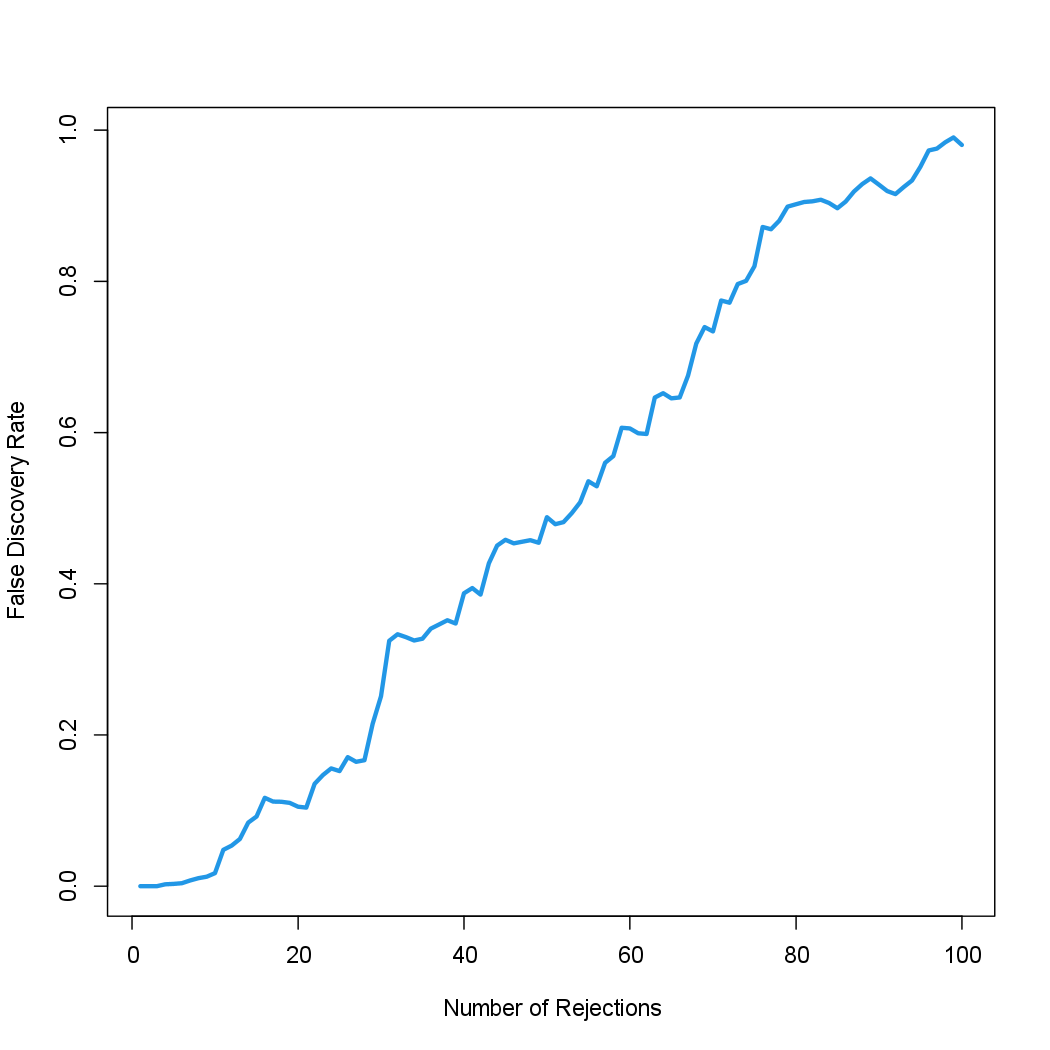
Now, for any given FDR, we can find the genes that will be rejected. For
example, with the FDR controlled at 0.1, we reject 15 of the 100 null
hypotheses. On average, we would expect about one or two of these genes
(i.e. 10% of 15) to be false discoveries. At an FDR of 0.2, we can reject
the null hypothesis for 28 genes, of which we expect around six to be
false discoveries. The variable index is needed here since we restricted our
analysis to just 100 randomly-selected genes.
> max(Rs[FDRs <= .1])
[1] 15
> sort(index[abs(Ts) >= min(cs[FDRs < .1])])
[1] 29 465 501 554 573 729 733 1301 1317 1640 1646 1706 1799 1942 2159
> max(Rs[FDRs <= .2])
[1] 28
> sort(index[abs(Ts) >= min(cs[FDRs < .2])])
[1] 29 40 287 361 369 465 501 554 573 679 729 733 990 1069 1073
[16] 1301 1317 1414 1639 1640 1646 1706 1799 1826 1942 1974 2087 2159
The next line generates Figure 13.11, which is similar to Figure 13.9, except that it is based on only a subset of the genes.
plot(Rs, FDRs, xlab = "Number of Rejections", type = "l", ylab = "False Discovery Rate", col = 4, lwd = 3)
As noted in the chapter, much more efficient implementations of the resampling
approach to FDR calculation are available, using e.g. the samr
package in R.